2026.06.21 | RA Space
'26.6月3주차 (6/15 ~ 6/20) 의료기기업계 주요 뉴스
- 올해는 미국 FDA(식품의약국)와 글로벌 의료기기산업에 특별한 한 해다. FDA 설립 120주년이자 미국 의료기기 규제 근간이 된 1976년 의료기기법(Medical Device Amendments to the Federal Food, Drug, and Cosmetic Act·FD&C Act) 제정 50주년을 맞이했기 때문이다.
- FDA는 최근 기념 성명을 통해 지난 50년간의 성과를 돌아보며 향후 의료기기 규제 방향을 제시했다. 단순한 기념행사가 아닌 인공지능(AI)·디지털 헬스·정밀의료 시대를 앞둔 FDA의 미래 비전을 담은 선언문이다.
- FDA가 이번 50주년 메시지에서 강조한 또 하나의 핵심 메시지는 ‘제품 생애주기 관리’(Total Product Life Cycle·TPLC)이다. 과거에는 시판 허가 취득이 규제의 끝이라고 생각했다면 이제는 허가 이후 실제 의료현장에서 축적되는 실사용 데이터(Real-World Data· RWD)를 지속적으로 수집·평가하는 것이 더욱 중요해지고 있다. 더불어 실사용 근거(Real-World Evidence·RWE), 시판 후 조사, 환자보고 성과(Patient Reported Outcomes) 등이 규제 의사결정의 핵심 요소로 자리 잡고 있다.
- FDA 의료기기법 50주년은 단순히 미국의 과거를 돌아보는 행사가 아니다. 글로벌 의료기기산업의 향후 50년이 어떤 방향으로 발전해야 하는지 보여주는 이정표이다. 지난 반세기 동안 FDA가 구축해 온 ▲위험도 기반 규제 ▲생애주기 관리 ▲환자 중심 규제의 세 가지 원칙은 앞으로도 글로벌 의료기기 규제의 중심축이 될 가능성이 높다.
- 이는 우리나라에도 중요한 시사점을 제공한다. 최근 혁신 의료기기 지정제도, 선진입·후평가, 의료 AI 제품의 신속 시장 진입 정책 등이 추진되고 있지만 상대적으로 시판 후 데이터 활용 체계는 아직 걸음마 단계에 머물러 있다. 특히 AI 의료기기와 디지털 헬스 기술은 출시 이후에도 지속적으로 성능이 변화할 수 있기 때문에 허가 시점의 평가만으로는 충분하지 않다. 향후 건강보험 청구자료, 전자의무기록, 환자 경험 데이터 등을 활용한 실사용 근거 기반 평가 체계 고도화가 요구되는 이유다.
- 식약처는 디지털의료기기 제조 및 수입업체를 대상으로 6월 16일 한국의료기기산업협회에서 ‘디지털의료기기 임상평가 간담회’를 개최한 데 이어 오는 6월 30일 서울지방식품의약품안전청에서 ‘디지털의료기기 허가·심사 임상평가 업무설명회’를 연다.
- 최근 시장 신속 진입을 위한 임상평가 제도가 시행됨에 따라 임상문헌을 포함한 평가보고서로 허가받은 디지털의료기기는 신의료기술평가가 유예되는 등 진입 절차가 대폭 개선됐다.
- 이에 맞춰 식약처는 간담회와 설명회를 통해 가이드라인 기반의 임상평가보고서 작성 방법과 심사 기준을 상세히 안내할 예정이다.
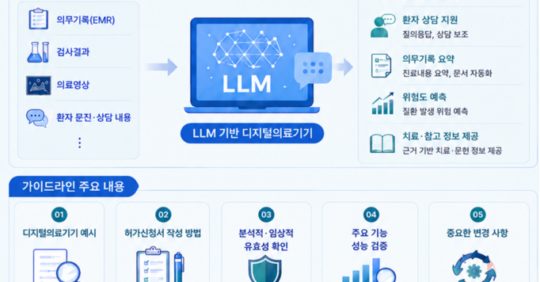
- 이어 업무설명회에서는 신기술의료기기 허가·심사 혁신 방안과 인공지능(AI) 디지털의료기기 변경관리계획서 작성법, 거대언어모델(LLM) 기반 생성형 인공지능 허가 가이드라인 등 최신 기준이 소개된다.
- 아울러 현장 참석이 어려운 업체를 고려해 오는 8월과 11월에 분기별 디지털의료기기 임상평가 온라인 교육도 병행해 실시하기로 한 식약처다.